

Introducing two powerful new capabilities in Azure Quantum Elements: Generative Chemistry and Accelerated DFT
Azure Quantum Elements is making research in chemistry and materials science faster, easier, and more productive by integrating new tools based on generative AI and high-performance computing.
Microsoft’s mission is to empower every person and every organization on the planet to achieve more. Azure Quantum Elements contributes to this mission by offering scientific capabilities based on AI and cloud high-performance computing (HPC). These user-friendly tools greatly increase the productivity of scientific research and remove barriers on the path to scientific discovery by substantially reducing the effort and expertise needed to perform what were previously daunting tasks. Specifically, these capabilities use Copilot for Azure Quantum, a natural-language interface that can be used by experts and non-experts alike, making Azure Quantum Elements accessible to more people and increasing the speed at which complex scientific problems can be solved.
Unlocking solutions to our most pressing challenges will require the world’s collective genius, and we’re excited to put new capabilities into the hands of scientists, students, and organizations such as Unilever so that everyone can contribute to making scientific discoveries that change the world for the better.
Since its launch, Azure Quantum Elements has served an important role in helping scientists achieve breakthroughs that have paved the way for more sustainable batteries and innovations in the pharmaceutical industry. Today, we’re announcing two new purpose-built capabilities in Azure Quantum Elements that will further increase the productivity and accessibility of chemistry and materials science research—Generative Chemistry and Accelerated DFT.
Generative Chemistry helps scientists discover novel, synthesizable, and useful molecules quickly
There are hundreds of millions of known molecular compounds and substances and many more remain to be discovered. In the field of chemistry, a major challenge is narrowing down an enormous number of candidate molecules to find the few that are best suited for a particular application. This challenge results in the streetlight effect, which is when the vast number of possibilities is reduced to a reasonable size, not based on the properties of the compounds, but by focusing only on those that have been studied previously. Relying on databases to identify suitable compounds limits the search space so that only known compounds are revealed as candidates for specific applications. Generative AI helps bring to light a much larger portion of the estimated 1060 possible combinations of atoms so that scientists are presented with novel candidates that are likely to serve the intended purpose.
Today, the Azure Quantum team at Microsoft is announcing Generative Chemistry—an upcoming capability that has the potential to greatly simplify this process and help scientists quickly discover and design new compounds with desired properties, thus increasing the productivity of product innovation.
Generative Chemistry, which will be available through Azure Quantum Elements private preview, is an end-to-end workflow that involves multiple steps:
- You provide information on desired molecular characteristics for your specific application. Additionally, you can supply reference molecules if you already have some possibilities in mind.
- The information you provide is used to generate seed molecules from a dataset—those seed molecules are then used to initiate the guided AI generation of candidate molecules for your application. This is accomplished by applying multiple AI models and a unique methodology to identify new compounds that match your criteria. In this step, you have several options for configuration such as selecting the most relevant generative AI model, specifying the number of molecules to be generated, indicating the key molecular properties of interest, and screening compounds for toxicity.
- AI-based screening models predict properties of the candidate molecules that are important for real-world applications—such as boiling point, density, or solubility. A feedback loop sends this information back to the guided AI generation (step 2) to modify the selection of candidate molecules. In this step, you also have the option to fine tune the AI models for your specific application.
- The pool of potential candidates is further narrowed through a step that uses AI-guided synthesis planning to determine the feasibility of making the molecules in a laboratory—this is important because some novel molecules with desired properties may be difficult to synthesize. In this step, synthesis pathways are predicted, and candidate molecules are filtered based on how easy they are to make.
- Highly accurate HPC simulations are performed on the top candidates. Accelerated DFT can be used to screen candidates for electronic properties such as dielectric constant, ionization potential, and polarizability. AutoRXN predicts chemical stability or reactivity, and it can also be used to provide insights on possible synthesis pathways.
- You are presented with the final candidate molecules from which you can select the most promising for laboratory synthesis and testing.
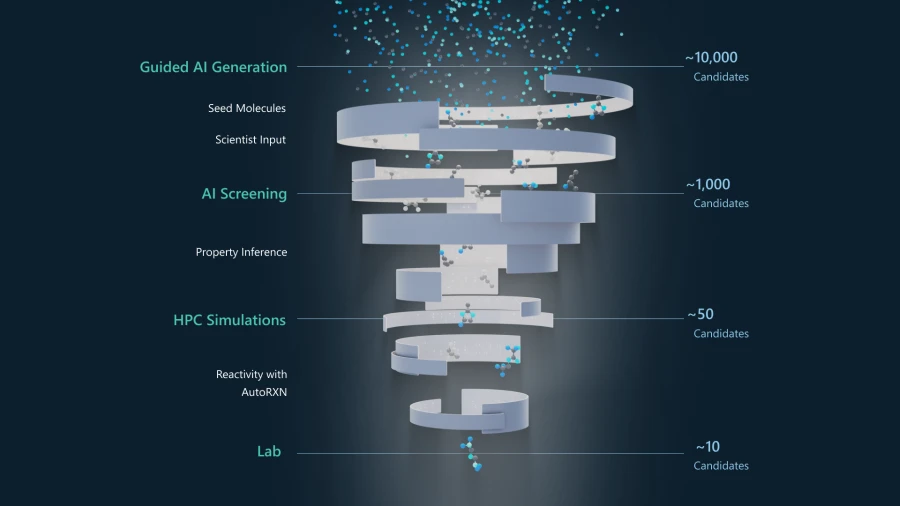
This entire process takes only days, shaving months or even years off trial-and-error laboratory experiments that were previously required to arrive at this point. Generative Chemistry suggests entirely new compounds and gives scientists the freedom to focus on only those molecules that are fit for the desired purpose—saving time, money, and effort. This new capability will allow for rapid progress in the development of novel therapeutics, sustainable materials, and more.

Accelerated DFT offers substantial increases in speed compared to other density functional theory codes
Density functional theory (DFT) is one of the most popular methods in computational chemistry due to its efficiency and accuracy in modeling quantum-mechanical properties. It allows researchers to simulate and study the electronic structures of atoms, molecules, and nanoparticles as well as surfaces and interfaces, thus predicting properties such as dielectric constant, ionization potential, and polarizability. Scientists can then tailor those properties to optimize them for specific applications.
Although DFT is highly valuable for research and product design, most DFT codes need to be run directly by the user in HPC clusters, which can be a difficult undertaking. Additionally, DFT—when run on traditional HPC hardware—requires substantial compute power and becomes constrained as the molecules being studied or designed increase in complexity and size.
To simplify and improve this process, Azure Quantum and Microsoft Research designed and launched Accelerated DFT, a code used to simulate the electronic structure of molecules. Accelerated DFT can determine the properties of molecules with thousands of atoms in a matter of hours. It performs substantially faster than other DFT codes and offers a 20-fold average increase in speed compared to PySCF, a widely used open-source DFT code.1
The setup of Accelerated DFT is made simple by providing the software as a service, requiring no code compilation or configuration on the user’s part, and it has a simplified API to streamline the computational process. Furthermore, a Python Software Development Kit (SDK) offers seamless integration into a wide variety of computational chemistry environments, which allows researchers to incorporate DFT calculations into complex chemistry workloads. Accelerated DFT is now available in Azure Quantum Elements private preview; it will also become part of Generative Chemistry.
Accelerated DFT can substantially expedite research across a wide spectrum of chemical disciplines by employing the power of Azure’s cloud architecture. Accelerated DFT can also produce large and highly accurate datasets of molecular properties that can be used to improve AI models, which require large amounts of training data. By rapidly generating training data, new molecules can be discovered faster, and known molecules can be improved, leading to innovations in therapeutics, sustainable products, and more.
“We have been very impressed by Accelerated DFT for simulating large systems with range-separated hybrid density functionals, where it speeds up the calculations considerably. Furthermore, the team at Microsoft has been very accommodating in quickly resolving technical challenges for our calculations of an active site of an enzyme.”—Tejs Vegge, Professor, Head of Section for Autonomous Materials Discovery, DTU Energy, Technical University of Denmark.
“Accelerated DFT has an easy-to-use Python interface, and the acceleration in calculations allows for the efficient use of novel hybrid functionals and a large basis set. Estimating key thermodynamic properties can thus be done in a matter of hours.
AspenTech is excited to be partnering with Microsoft to evaluate the application of quantum chemistry in our physical properties system for process modeling, especially in our sustainability pathways solutions. We anticipate that working with Accelerated DFT will result in strong use cases for characterizing new molecules and materials that are crucial to decarbonizing and circularizing industrial and consumer materials.”—Heiko Claussen, Senior Vice President, Artificial Intelligence Technology at AspenTech.
Azure Quantum Elements incorporates quantum computing
With the addition of each new feature, Azure Quantum Elements becomes a more effective tool for scientists by harnessing AI, HPC, and emerging hybrid-computing capabilities that bring the power of quantum computing to scientific challenges. Recently, we simulated a chemical catalyst by combining classical supercomputers, AI, and logical qubits created with Microsoft’s qubit-virtualization system and Quantinuum’s H1 hardware. In the coming months, we will introduce advanced logical qubit capabilities from Microsoft and Quantinuum to the private preview of Azure Quantum Elements. This classical-quantum hybrid computing offering follows our quantum computing milestone with Quantinuum in which we created the most reliable logical qubits on record with an error rate 800 times better than that of the corresponding physical qubits.
Advances in AI and quantum computing have the potential to help researchers solve global scientific challenges. In the future, we plan to offer a quantum supercomputer that can simulate interactions of molecules and atoms at the quantum level, which is beyond the reach of classical computers. This capability is expected to transform research and innovation across many industries. To advance the safe use of these technologies, we will ensure that they are developed and deployed responsibly. We will continue to adopt thoughtful safeguards, building on our commitments to responsible AI and embracing responsible computing practices as these capabilities grow.
Learn more about how Azure Quantum Elements is revolutionizing chemistry and materials science
- Read about our collaboration with Unilever in the Official Microsoft Blog.
- Explore the product overview of Azure Quantum Elements.
1 Details on the performance of the private preview of Accelerated DFT can be found in the preprint of Acceleration without Disruption: DFT Software as a Service.

